Real Data with Simulated Signals: Part I
Lei Sun
2017-05-18
Last updated: 2017-12-21
Code version: 6e42447
library(ashr)
library(edgeR)
library(limma)
library(qvalue)
library(seqgendiff)
library(sva)
library(cate)source("../code/gdash.R")Introduction
Using David’s package seqgendiff, we are adding artefactual signals to the real GTEx Liver RNA-seq data.
mat = read.csv("../data/liver.csv")The true signal comes from a mixture distribution
\[
g\left(\beta\right) = \pi_0\delta_0 + \left(1 - \pi_0\right)N\left(0, \sigma^2\right)
\] The simulated data matrices are then fed into edgeR, limma pipeline. In the following simulations, we always use \(5\) vs \(5\).
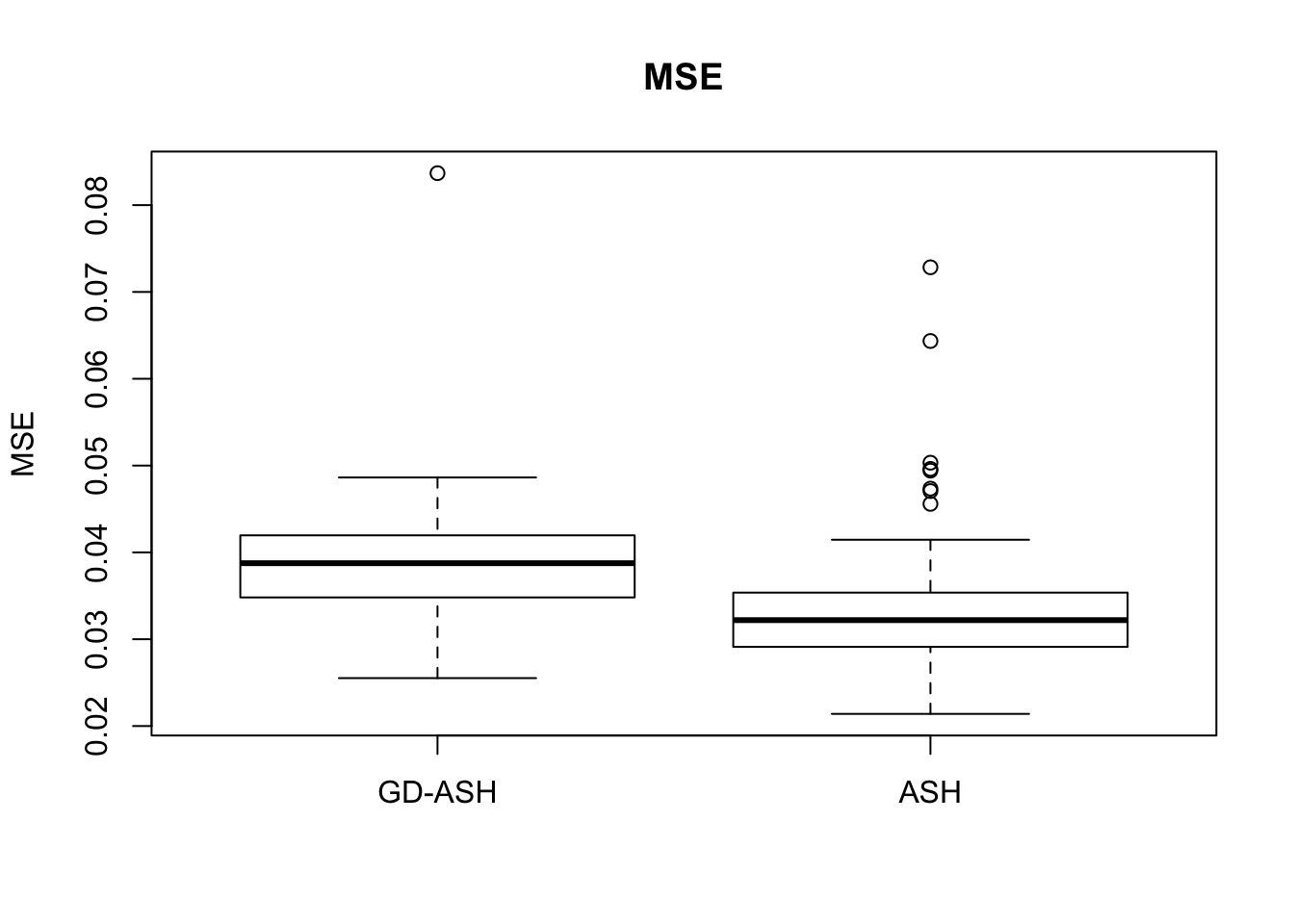
Case 1: \(\pi_0 = 0.9\), \(\sigma^2 = 1\).
N = 100
nsamp = 10
pi0 = 0.9
sd = 1
system.time(ashvgdash <- N_simulations(N, mat, nsamp, pi0, sd)) user system elapsed
6854.603 627.845 12855.081 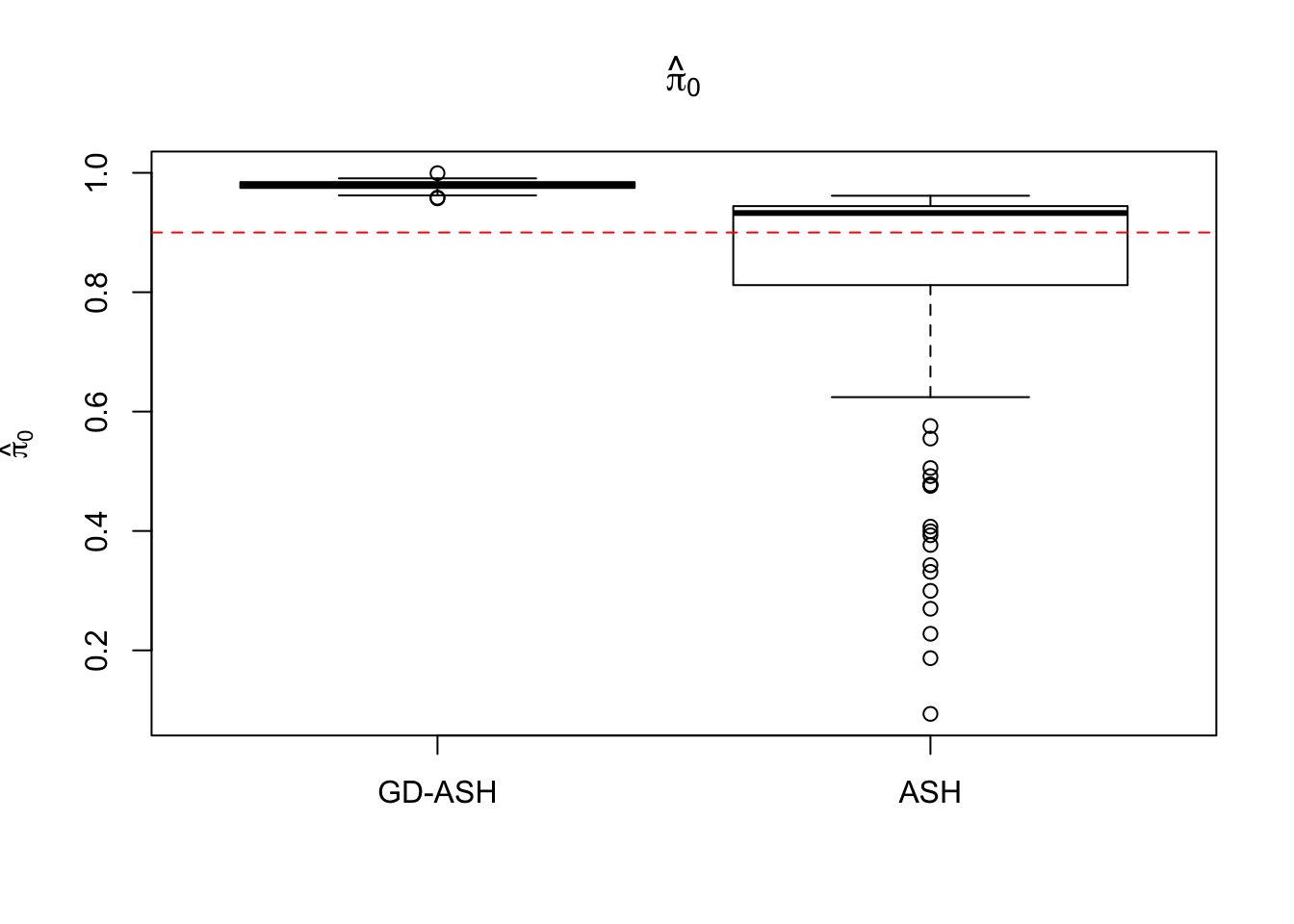
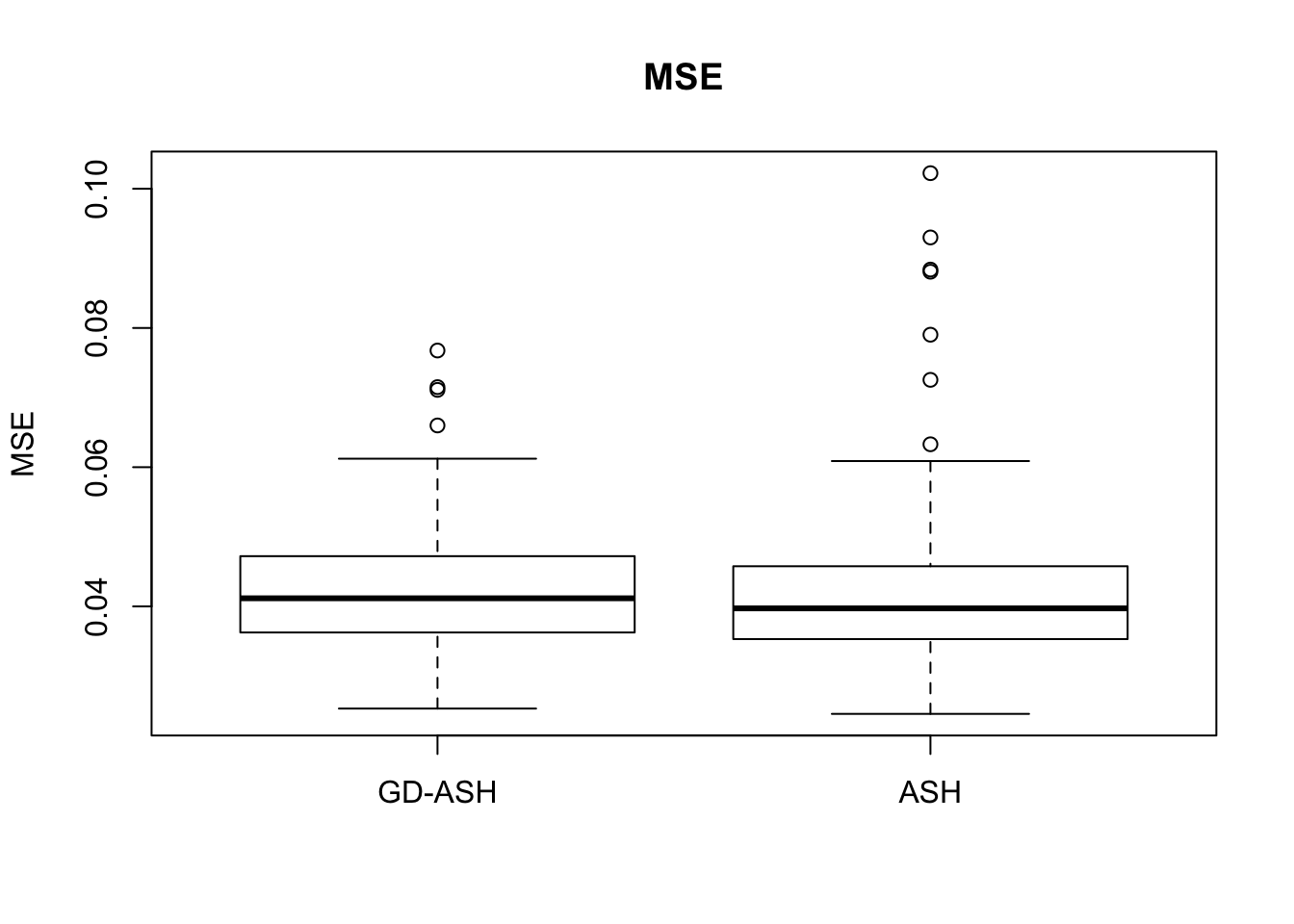
Case 2: \(\pi_0 = 0.9\), \(\sigma^2 = 4\)
N = 100
nsamp = 10
pi0 = 0.9
sd = 2
system.time(ashvgdash <- N_simulations(N, mat, nsamp, pi0, sd)) user system elapsed
5877.223 621.433 5256.693 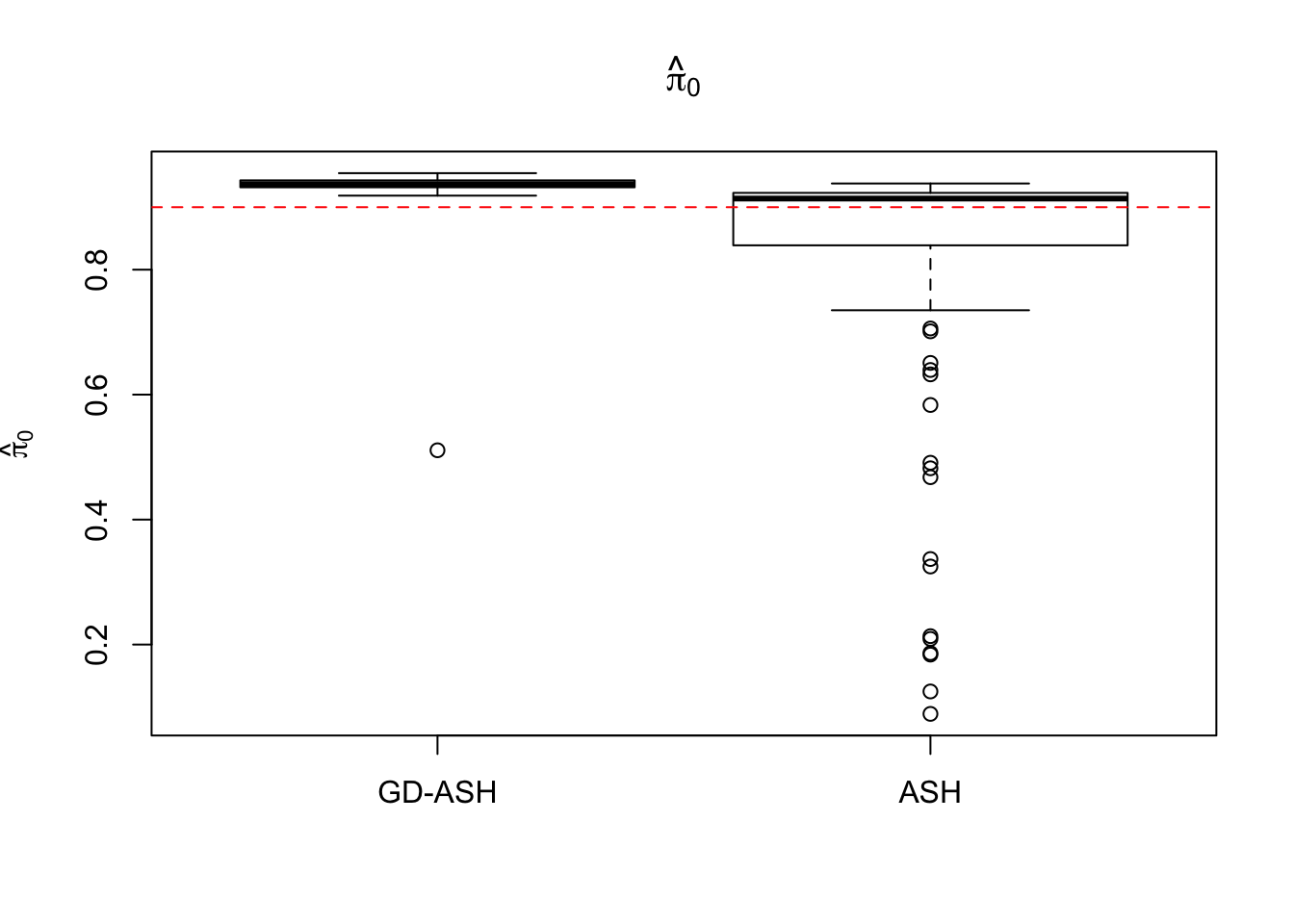
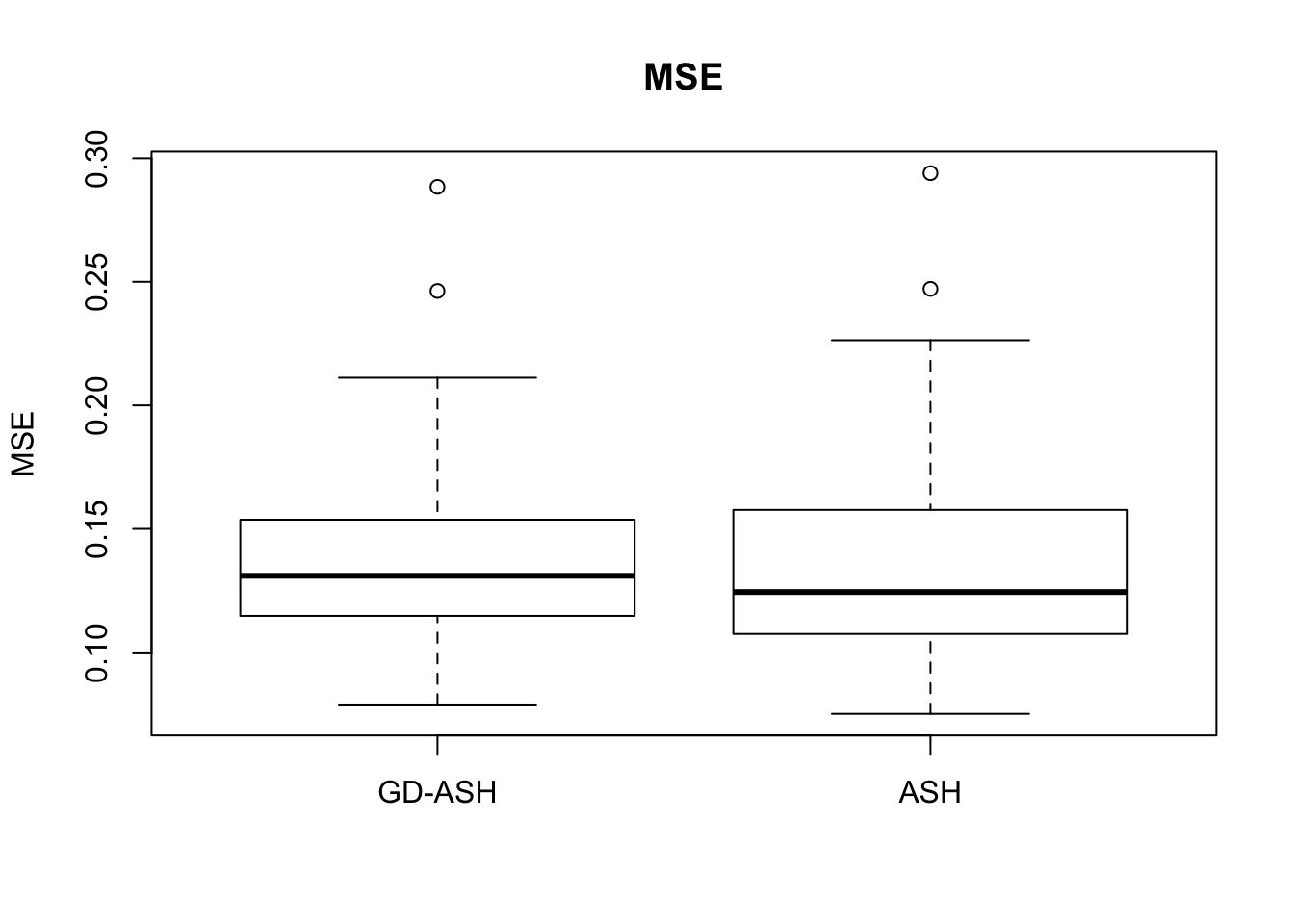
Case 3: \(\pi_0 = 0.5\), \(\sigma^2 = 4\)
N = 100
nsamp = 10
pi0 = 0.5
sd = 2
system.time(ashvgdash <- N_simulations(N, mat, nsamp, pi0, sd)) user system elapsed
5269.488 574.588 15042.479 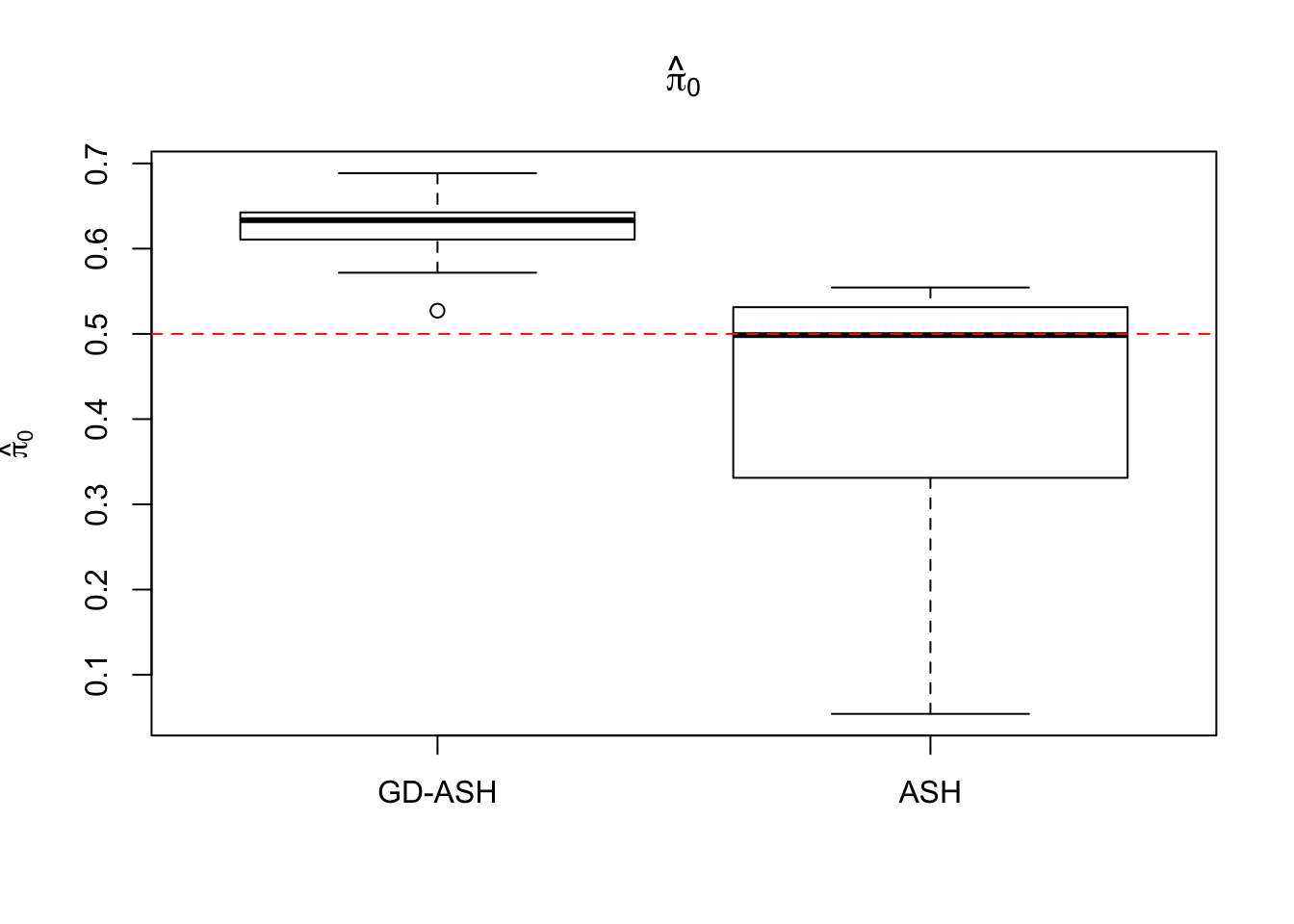
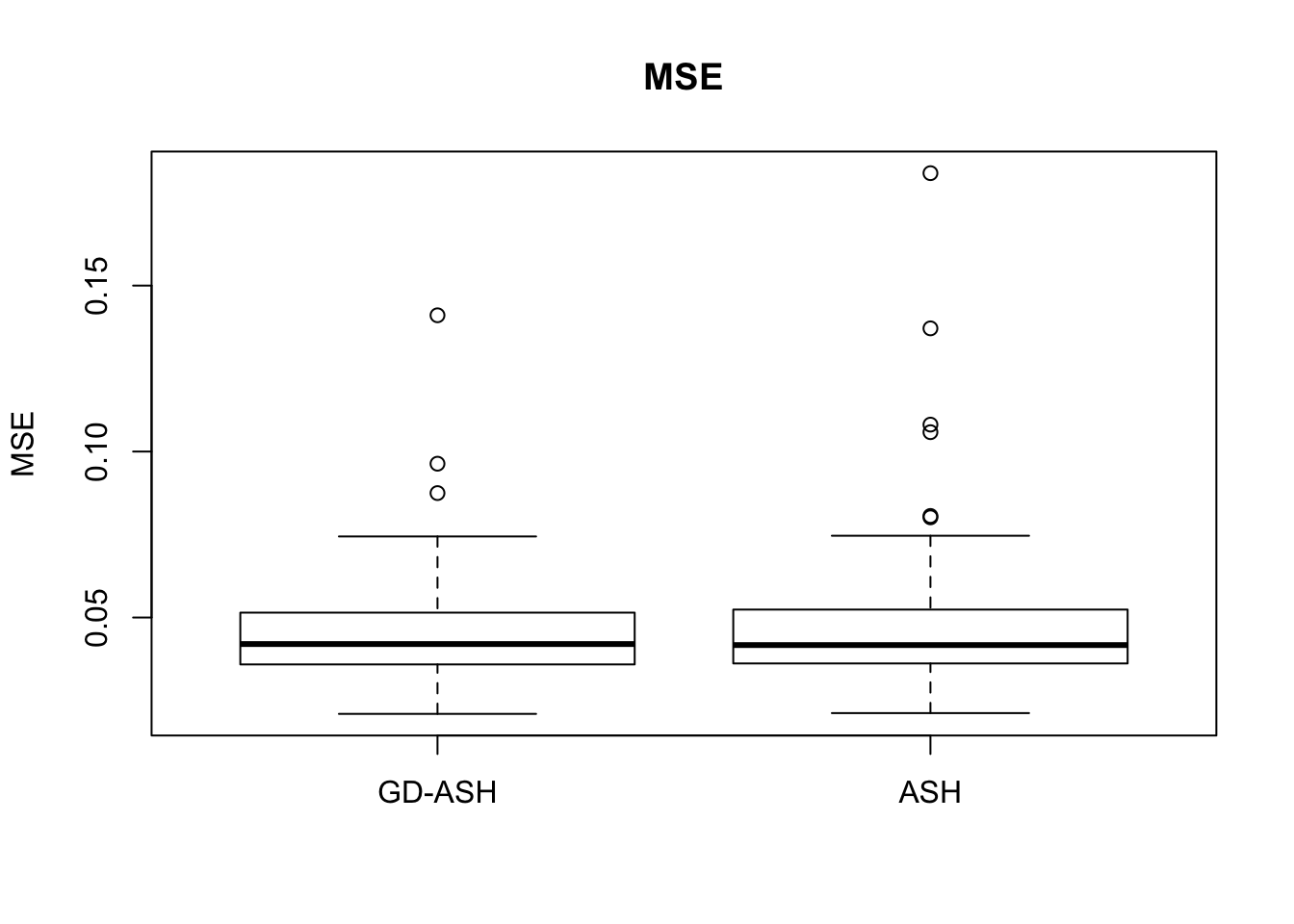
Case 4: \(\pi_0 = 0.9\), \(\sigma^2 = 9\)
N = 100
nsamp = 10
pi0 = 0.9
sd = 3
system.time(ashvgdash <- N_simulations(N, mat, nsamp, pi0, sd)) user system elapsed
5321.625 558.205 15356.345 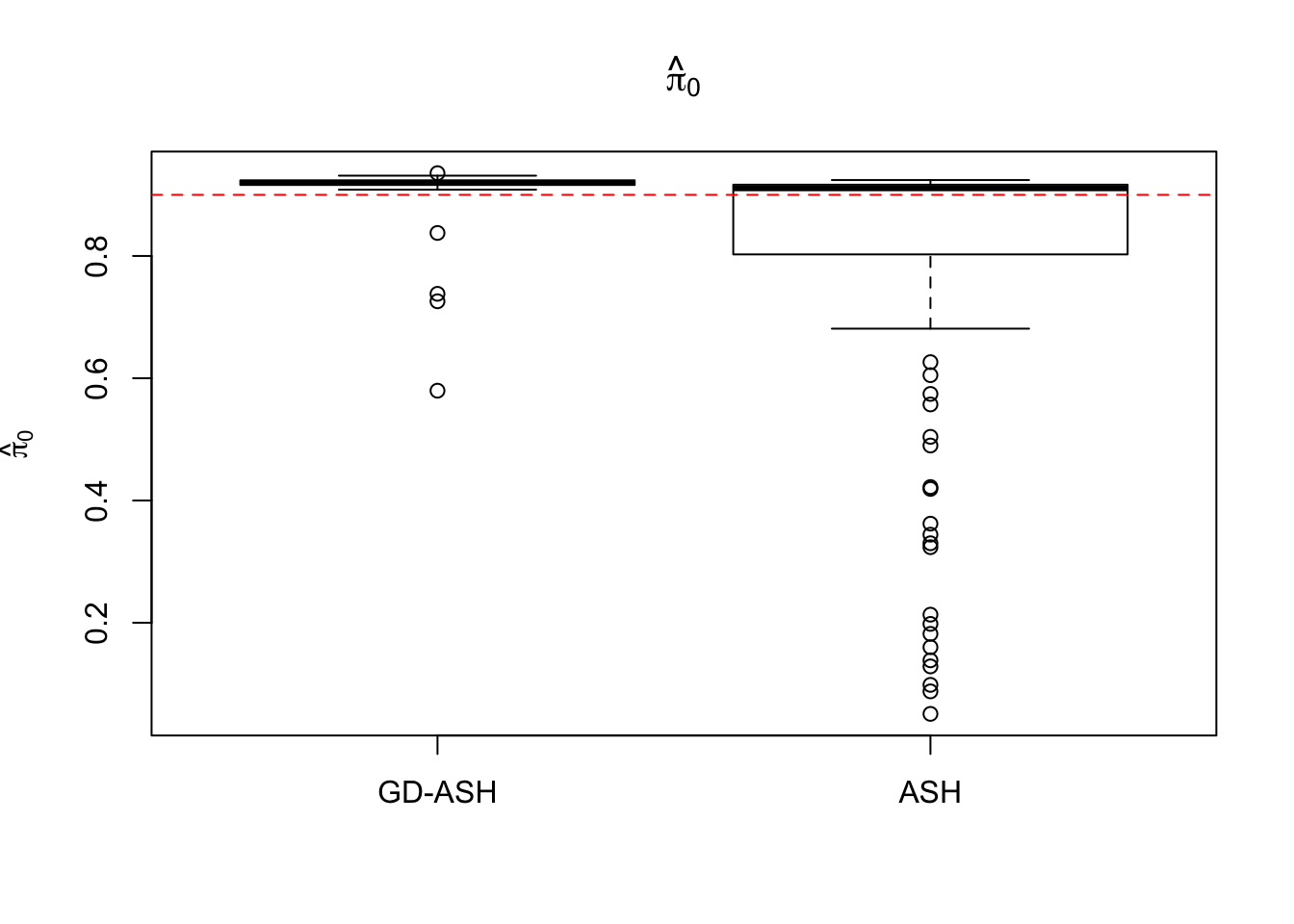
Session information
sessionInfo()R version 3.4.3 (2017-11-30)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS High Sierra 10.13.2
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] Rmosek_8.0.69 PolynomF_1.0-1 CVXR_0.94-4
[4] REBayes_1.2 Matrix_1.2-12 SQUAREM_2017.10-1
[7] EQL_1.0-0 ttutils_1.0-1 cate_1.0.4
[10] sva_3.26.0 BiocParallel_1.12.0 genefilter_1.60.0
[13] mgcv_1.8-22 nlme_3.1-131 seqgendiff_0.1.0
[16] qvalue_2.10.0 edgeR_3.20.2 limma_3.34.4
[19] ashr_2.2-2
loaded via a namespace (and not attached):
[1] Biobase_2.38.0 svd_0.4.1 bit64_0.9-7
[4] splines_3.4.3 foreach_1.4.4 ECOSolveR_0.3-2
[7] R.utils_2.6.0 stats4_3.4.3 blob_1.1.0
[10] yaml_2.1.16 RSQLite_2.0 backports_1.1.2
[13] lattice_0.20-35 digest_0.6.13 colorspace_1.3-2
[16] R.oo_1.21.0 htmltools_0.3.6 plyr_1.8.4
[19] XML_3.98-1.9 esaBcv_1.2.1 xtable_1.8-2
[22] corpcor_1.6.9 scales_0.5.0 scs_1.1-1
[25] git2r_0.20.0 tibble_1.3.4 annotate_1.56.1
[28] gmp_0.5-13.1 IRanges_2.12.0 ggplot2_2.2.1
[31] BiocGenerics_0.24.0 lazyeval_0.2.1 Rmpfr_0.6-1
[34] survival_2.41-3 magrittr_1.5 memoise_1.1.0
[37] evaluate_0.10.1 R.methodsS3_1.7.1 doParallel_1.0.11
[40] MASS_7.3-47 truncnorm_1.0-7 tools_3.4.3
[43] matrixStats_0.52.2 stringr_1.2.0 S4Vectors_0.16.0
[46] munsell_0.4.3 locfit_1.5-9.1 AnnotationDbi_1.40.0
[49] compiler_3.4.3 rlang_0.1.4 grid_3.4.3
[52] leapp_1.2 RCurl_1.95-4.8 iterators_1.0.9
[55] bitops_1.0-6 rmarkdown_1.8 gtable_0.2.0
[58] codetools_0.2-15 DBI_0.7 R6_2.2.2
[61] reshape2_1.4.3 ruv_0.9.6 knitr_1.17
[64] bit_1.1-12 rprojroot_1.3-1 stringi_1.1.6
[67] pscl_1.5.2 parallel_3.4.3 Rcpp_0.12.14 This R Markdown site was created with workflowr